药物体内过程
| A+医学百科 >> 药物体内过程 |
药物体内过程(drug movement in the body),药物在机体内的吸收、分布、代谢和排泄过程。在现代药理学研究中,常把吸收和分布称为处置,而将代谢和排泄称为消除。药物代谢也称药物的生物转化,而吸收、分布和排泄称为药物的转运。定量地研究药物体内过程动态规律的科学称为药物代谢动力学,简称药代动力学。药代动力学研究机体对药物的作用规律,是药理学的内容之一(药理学的另一个内容是药效学,即研究药物对机体作用规律的科学)。现代研究证明,药物疗效及毒副反应的强度和持续性与药物体内过程密切相关。因此,药代动力学研究对于推动新药的设计与开发以及提高药物治疗学水平有着极为重要的意义。
药物进入机体以后,其作用经历着增长-平衡-消除的变化过程。这种过程反映着药物的吸收、分布、代谢和排泄随时间变化的过程,即药物的时间过程,这个过程可以用血浆药物浓度-时间曲线(简称药-时曲线)加以描述。
目录 |
药-时曲线
以时间为横坐标,以药物的某些数量特征(如血药浓度、尿药浓度)为纵坐标所做的曲线。借助于这些曲线可以分析并阐明药物的动力学特征。目前采用较多的是血浆药物浓度-时间曲线(图1)。这是因为多数药物的药理效应强度和持续性与其在作用部位的药物浓变化度密切相关,而药物在血液中的浓度变化一般可成比例地反映其在作用部位的变化;收集血标本比较方便,而且先进的分析测试方法可以用极少标本测得微量药物浓度。此外,也可用尿液、唾液等其他标本进行研究。
模拟药物体内过程的数学模型
为了研究药物在体内转运的量变规律,药代动力学分析采用数学模型模拟药物的体内过程,线性乳突型模型是应用较广、研究较多的模型。这种模型抽象地将机体看作一个系统,再根据药物转运的特征将系统划分为一个或几个房室,从而得到一室或多室模型。常用的有一室模型、二室模型和三室模型。
一室模型
将机体视为一个均匀的系统,药物进入机体即迅速分布,瞬间达到平衡。在动力学处理中,不考虑分布问题,药物只是从体内消除。这种模型适用于在体内迅速分布的药物,其特点是简单,但对大部分药物体内过程的分析不够精确。
多室模型
将机体划分为中央室和外周室。多数药物进入机体后,需要一定时间才能在全身分布完全。由于药物在不同体液和组织器官中转运的速率不同,在动力学处理中,将血液和血流丰富、药物容易进入并迅速与血中药物达到平衡的组织器官如心、肝、脑、肾、肺等归为中央室;将血流量少、药物不易进入并较慢与血液药物达到平衡的组织器官如静止状态的肌肉、脂肪等归为外周室。药物在中央室与外周室之间按一级速率转运,处于动态平衡中。只有一个外周室的模型称二室模型;有些药物在外周室的组织器官中转运速率有较大差异,因而分为两个外周室,称为三室模型。其转运速率常数分别为K12和K21。应当指出,房室的划分不是固定的,而是取决于组织器官的血流特征、对药物的摄取能力及药物本身的性质,例如对一种脂溶性药物,脑属于中央室;而对一种极性大的药物,脑则可以属于外周室。多室模型既考虑药物的分布,又考虑其消除,比较全面。
动力学类型
药物在体内的转运是一个随时间而变化的动力学过程。在研究化学反应动力学时,从反应速度与反应物的量(或浓度)之间的关系出发,将反应分为零级、一级或多级反应。在药代动力学研究中也引入了这种概念。
一级动力学
即一级速率过程,又称线性动力学。其特点是药物的转运(转运到其他部位或消除掉)速率与该部位药物的量(或浓度)的一次方成正比。例如,一级消除动力学指血中药物的消除速率与血药浓度的一次方(即血药浓度)成正比。血药浓度高,则消除速率高,即单位时间内消除的药量多;血药浓度降低,药物的消除速率也下降。
零级动力学
即零级速率过程。其特点是药物的转运速率与该部位药量(或浓度)的零次方成正比,即为恒定的,与所在部位的药量(或浓度)无关。零级速率过程多数情况下是因药量过大而超过机体最大处理能力所致。
非线性动力学
在治疗剂量时,大多数药物的药代动力学符合一级速率过程。但也有些药物如乙醇和苯妥英钠等,在大剂量时其体内过程出现非线性动力学过程。因为药物在体内的某些过程,如药物代谢或经肾小管分泌而转运的过程中,通常有酶或载体系统参与,这些系统具有一定的容量限度。当大剂量时,消除过程出现饱和现象。低浓度时,符合线性过程。
吸收
药物自给药部位进入血液循环的过程。除直接注入血管者外,一般给药方法都要经过吸收过程。
吸收的途径
皮下或肌肉注射给药通过毛细血管壁吸收,一般吸收快速而完全。口服给药通过胃肠粘膜吸收,虽弱酸性药物可在胃中吸收,但大部分仍在肠中吸收。药物在胃肠吸收的途径主要是经过毛细血管进入肝门静脉。某些药物在通过肠粘膜及肝脏灭活代谢后,进入体循环的药量减少,这种作用称为首过效应。经淋巴吸收的药物较少。舌下含锭、经肛灌肠及栓剂由于接触面小,吸收量较口服的少;但由于不经肝门静脉,药物破坏少,作用较快。挥发性药物和气体如乙醚和亚硝酸异戊酯等,经肺泡吸收,速度快。除少数脂溶性极大的有机溶剂、有机磷酸酯等外,皮肤对大多数药物不吸收。
影响吸收的因素
影响药物吸收的因素一方面来自药物,包括药物的物理化学性质、剂型及对组织的亲和力等;另一方面来自机体,包括胃肠蠕动情况、胃内容物、胃排空速度及注射部位的血流情况等。
生物利用度
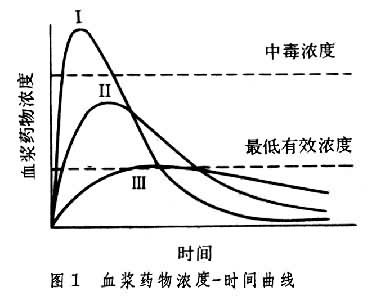
用以描述药物吸收进入血液循环的量和速度的概念,又称全身利用度。评定药物的生物利用度用三项药代动力学参数,即峰浓度、峰时间和药-时曲线下面积 (AUC)。峰浓度指药物在血液中达到的最高浓度;峰时间指达到最高浓度的时间;AUC指药物进入血液循环后至全部原型药物排出体外过程中药-时曲线下的面积。峰浓度和峰时间衡量药物被吸收利用的速度,而AUC描述药物吸收利用的程度。图1为口服同一药物相同剂量的不同制剂得到的三条药-时曲线,三种制剂被吸收的速度明显不同,制剂Ⅰ吸收太快以致峰浓度达到中毒浓度;制剂Ⅲ则吸收太慢,峰浓度处于有效浓度之下;而制剂Ⅱ的吸收居于前二者之间,峰浓度在有效浓度范围内。三者的吸收速度不同,但AUC相同,表明其被吸收利用的程度相同。
生物利用度对临床治疗具有重要意义。以阿司匹林为例,当治疗的目的是快速止疼时,宜选用曲线Ⅱ的制剂,如水溶性阿司匹林。对于风湿性关节炎的长期用药治疗则宜选用曲线Ⅲ的制剂如肠溶阿司匹林,因为该制剂虽单次给药时未达有效浓度,但多次给药后其蓄积浓度可达有效浓度且维持时间长。有时曲线Ⅰ的制剂也具治疗意义,如舌下给三硝酸甘油,大量快速吸收可使心绞痛症状迅速缓解,但也会因达中毒浓度而导致副反应,如头痛。
分布
药物吸收后首先进入血液循环,然后向机体有关部位转运的过程。药物在体内的分布多数是不均匀的,且处于动态平衡中,随着其吸收和消除不断变化着。药物在全身分布的规律决定着药物在靶器官(作用器官)的浓度,从而决定着其药理作用的强度及持续时间。
影响药物分布的因素
大致有以下几方面:
①药物的物理化学性质。主要包括分子大小、脂溶性、解离度、酸碱性、药物与组织的亲和力及稳定性等,均影响药物的分布。
②局部组织器官血流量。药物在组织器官中分布达到平衡的速度主要取决于通过该组织器官的血流速度。通常心、肺、脑、肝、肾等血流较快,分布达到平衡较快;肌肉次之;脂肪组织很慢。根据药物在不同组织器官中分布速度的差异情况可将机体视为一室或多室模型。
③与血浆蛋白的结合。药物进入血液后,或多或少地与血浆蛋白结合。结合型药物失去活性。由于药物与血浆蛋白结合,使血中游离药物浓度下降,有利于继续吸收;结合后的药物不易穿透毛细血管壁、各种细胞膜屏障及肾小球,可限制其进一步转运,减慢消除。药物的血浆蛋白结合产物是疏松的、可逆的,与游离型(未与蛋白结合者)处于动态平衡中,因而是一种在体内的暂时储存形式。血浆蛋白与药物的结合具有一定的限度,达到饱和后继续增加剂量会导致游离药物浓度迅速升高而引起中毒。临床用药时要考虑药物血浆蛋白结合的情况。
④细胞膜屏障。它是影响药物分布的重要因素,如血脑屏障和胎盘屏障。以血脑屏障为例,许多分子较大、极性较强的药物分子不能穿过血脑屏障进入脑组织。当药物与血浆蛋白结合后分子变大也不能穿过血脑屏障。磺胺噻唑(ST)与血浆蛋白结合多,透过血脑屏障进入脑脊液少,而磺胺嘧啶(SD)与血浆蛋白结合少,进入脑脊液多,故治疗流行性脑脊髓膜炎时应选用SD。
此外,机体的病理状况及合并用药等都可影响药物的分布。
表观分布容积(Vd)
反映药物在体内分布程度的药代动力学参数。Vd是体内药量与血药浓度的比值。Vd是假定药物在体内均匀分布情况(即各组织浓度与血浓度相等)下的分布容积。加“表观”二字,是因为这个容积并不等于机体中真正的容积数值。虽然有些药物的Vd值与已知的体液容积相似,但也有许多药物的Vd值大于或远大于总体液量,如心得安的Vd值大药为 3L/kg体重(体液约为0.6L/kg体重)。药物的Vd值越大,表明药物在组织中分布越广泛。利用Vd值,可根据血浆浓度算出体内药量,也可以估算欲达到某个血药浓度应选用的剂量或用一定剂量后某一时间的血药浓度,从而制订合理的给药方案。
代谢
药物在体内发生结构转化的过程,又称生物转化。大多数药物代谢发生在肝脏,有的也发生在其他部位,如血浆、肾脏等。尽管有些药物在代谢过程中产生具有药理活性的物质,但代谢的最终结果是使药物失去药理活性(灭活)。药物代谢有三种情况:①非活性物质代谢为活性物质。有些不具药理活性的物质在体内代谢后产生具有药理活性的物质,前者称为前药。如羧苄苯青霉素代谢为羧苄青霉素而起作用,前者的吸收好于后者。多巴胺不能进入脑内,因此对帕金森氏病无效,而左旋多巴易进入脑内并代谢为多巴胺发挥疗效。②一种药物代谢为另一种药物。某些药物代谢为与其作用相似或更强的药物,如海洛因代谢为吗啡,非那西汀代谢为扑热息痛。有些药物代谢为毒性物质,如异烟肼代谢为具有肝毒性的乙酰化产物。③代谢为无活性物质。大多数药物的代谢属于此类。
药物代谢分两个阶段。第一阶段为氧化、还原或水解过程,第二阶段为结合过程。各种药物在体内的代谢过程不同,或经第一阶段,或经第二阶段,或二者均有。代谢的最终产物比原型增加极性和水溶性以便排出体外。各种药物经代谢部分的多少(即代谢部分占总药量的百分数)不同,有些药物不经代谢而直接从体内消除。
药物代谢靠酶的促进,主要是肝脏微粒体混合功能氧化酶系统(简称肝药酶),其中主要的氧化酶是细胞色素P-450。肝药酶的作用特异性很差,许多药物都经它代谢,而且其活性有限,因此药物之间存在竞争性抑制作用。肝药酶的活性易受多种药物的影响发生变化。一类药物可诱导肝药酶活性提高,加速药物代谢,这类药物称为肝药酶诱导剂;另一类药物能抑制肝药酶,使其活性降低,减慢药物代谢,这类药物称为肝药酶抑制剂。合并用药时要注意药酶诱导剂或抑制剂的作用,避免由此引起的毒副反应;也可利用其相互作用改善治疗。
影响药物代谢的因素有遗传、年龄、肝血流量、肝脏病理状况及其他药物的存在等。
排泄
药物在体内的最后过程。排泄的主要途径是肾脏,还有胆、肺、唾液腺及汗腺等。多数药物经代谢后变为极性大的化合物排出体外,也有些药物以原型排出体外,或部分以原型、部分以代谢物排泄。
肾脏排泄
经三种过程:肾小球过滤、肾小管被动重吸收和肾小管主动分泌。除与血浆蛋白结合的药物外,游离的药物及其代谢物都通过肾小球过滤进入肾小管,进入肾小管后,脂溶性大的药物随着浓度的增加可被肾小管重吸收进入血液循环,使肾清除率减慢。这类药物的肾清除率受尿pH与尿液流速的影响。极性高、水溶性大的药能顺序通过肾小管排泄。肾小管主动分泌的药物一般排泄较快。经肾小管分泌的药物有弱酸性药物青霉素、速尿、丙磺舒和尿酸等与弱碱性药物苯丙胺和奎宁等。经肾小管分泌的同类药物之间具有竞争性抑制作用,如丙磺舒抑制青霉素的排泄使其半衰期延长,药理作用增强。药物经肾脏浓缩后可达尿中浓度很高,这是某些药物治疗泌尿道感染的依据,但也可由此导致肾毒性。
胆汁排泄
某些药物经肝脏代谢后向胆汁分泌。这些药物自胆汁排泄百分比很大,且胆道内浓度很高,有利于肝胆系统疾患的治疗。这类药物有利福平、四环素和红霉素等。自胆汁排进十二指肠的结合型药物在肠中经水解后被再吸收,形成肝肠循环,使药物的作用明显延长。
药物的代谢和排泄统称为药物的消除过程。用以描述消除过程的重要药代动力学参数是半衰期 (t1/2)和清除率(CL)。半衰期是衡量药物从体内消除速度的动力学参数,指血浆中药物浓度下降一半所需要的时间,以时间为单位,如天、小时和分等。t1/2不仅提供关于药物在体内停留时间的直观概念,它的实用价值还在于帮助人们确定重复给药时适当的时间间隔以及计算多剂给药中达到稳态浓度所需要的时间。清除率是机体消除药物速率的另一种表示方法,指单位时间内有多大容积血浆所含的药物被清除。单位是升/小时(L/h)或毫升/分(ml/min)。清除率是设计给药方案的最重要参数之一。根据药物的清除率和生物利用度,可以通过调整剂量和给药间隔建立和维持所需要的稳态血药浓度。
参看
| 关于“药物体内过程”的留言: | |
|
目前暂无留言 | |
| 添加留言 | |