临床生物化学/先天性代谢缺陷病
| 医学电子书 >> 《临床生物化学》 >> 遗传性疾病的生物化学与分子生物学诊断 >> 常见遗传性疾病的发生与基因诊断 >> 先天性代谢缺陷病 |
| 临床生物化学 |
|
|
(一)苯丙酮尿症
苯丙酮尿症(PKU)因患者尿中含大量苯丙酮酸而得名病因是患者肝缺乏苯丙氨酸羟化酶,使由食物摄入体内的苯丙酮酸不能正常代谢为酪氨酸,导致血清中苯丙氨酸浓度升高,可高达50-100mg/dl(正常参考值为1-3mg/dl)。大量的苯丙氨酸使旁路代谢活跃,经苯丙氨酸转氨酶作用生成苯丙酮酸。
苯丙氨酸羟化酶是在肝细胞中合成的,即在肝细胞中专一表达而在胎儿的绒毛细胞或羊水细胞中并不表达,给PKU的产前诊断带来了困难。
由于诊断分子生物学技术的发展,1983年胡流清等人完成了人苯丙氨酸羟化酶cDAN探针的制备。他们利用探针和Southern blot技术进行分子杂交,通过限制性片段长度多态性(restriction fragment-length polymerphism ,RFLP)分析实现了PKU的产前基因诊断。他们先用MSP-I限制性内切酶消化正常人基因组DNA,经核酸电泳后,将DNA片段转移到一张硝酸纤维膜(或尼龙膜)上,再用32p标记的人苯丙氨酸羟化酶cDNA探针杂交后,再经放射自显影技术显示正常人群有23kb和19kb两种限制性片段长度。同时他们调查了一个PKU家系,进行比较发现,同样用Msp-I内切酶消化,父母同时有23kb和19kb DNA,患儿只有19kb片段。说明在该家系中,苯丙氨酸羟化酶突变基因是与19kb片段连锁的。连锁简而言之就是指同一条染色体上的基因联合遗传的现象。因此在该家系中的第2胎,如果只出现19kb DNA片段将是患儿,而同时出现23kb和19kb DNA片段或只出现23kbDNA片段的都是正常个体,而且只出现23kb DNA片段者不携带突变基因,不是突变基因的携带者。正常人出现19kb仅代表蛋白质和酶的多态。PKU家系的患儿为19kb,正常儿为23kb说明该PKU家系的基因突变与19kb连锁。(表15-2)
表15-2 人苯丙氨酸羧化酶基因核酸电泳与cDNA探针杂交自显影
| 正常人多态 | PKU家系 | |||||
| 父 | 母 | 患儿 | 正常胚胎 | |||
| 23kb | - | - | - | - | - | |
| 19kb | - | - | - | - | ||
1986年胡流清等进一步证明苯丙氨酸羧化酶的表达异常是基因点突变所致,并确定了突变点位置,并由此设计了正常和突变的各含21个核苷酸的寡核苷酸探针。
↓
正常探针5′TCCATTAACAGTAAGTAATTT3′
突变探针5′TCCATTAACAATAAGTAATTTT3′
↑
用这两个探针分别与PKU家系成员的DNA杂交,证实与正常探针杂交者为正常个体,与突变探针杂交者为患者,同时可与两个探针杂交者为正常个体,但携带突变基因。此法已成功用于产前诊断。(图15-1)
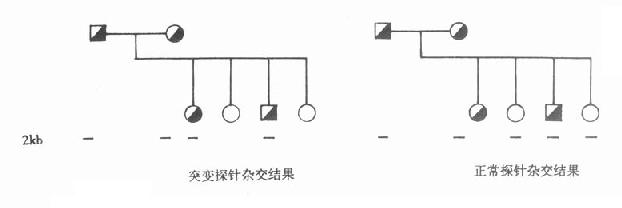
图15-1 正常与突变苯丙氨酸羟化酶探针与PKU家系成员的DNA杂交结果
杂交技术由于费时、操作复杂而难以在基层实验室推广。现在多用聚酶链反应(PCR)扩增技术进行基因诊断,或在PCR基础上再进行探针杂交,在提高灵敏度的同时又提高特异性,而且快速、经济、简便实用。
(二)嘌呤代谢紊乱与痛风
尿酸是嘌呤核苷酸分解代谢的重要产物,血和尿中尿酸浓度的检测是嘌呤代谢紊乱的重要化学指标。可由经典的磷钨酸还原法和高度特异的尿酸酶等化学方法检测。
健康成年男子每天约生成尿酸600-700mg,其中60%-70%经肾排出,剩余约200mg排入肠道由细菌降解。体内尿酸池维持在1200mg尿酸水平。肾排出尿酸的机制是由于尿酸分子量小(168u),可全部经肾小球滤过,但至少有98%被近曲小管重吸收,再经远曲小管主动分泌,因此随尿排出的尿酸主要由肾小管所分泌。肾每天约排出尿酸400-500mg(2.4-3.0mmol/L),相当于肾小球原滤液中含尿酸的4%-5%。
在血浆pH为7.4时,尿酸几乎完全以单钠尿酸盐的形式存在,溶解度有限,约为0.42mmol/L(7.0mg/dl);而尿酸的溶解度更低,在pH5的尿液中,比尿酸难溶20倍。健康成年男子的血清尿酸浓度约0.3mmol/L,女子约低20%。血清尿酸水平随年龄增加而增高,男性比女性更明显,可达0.46mmol/L。血清尿酸超过0.42mmol/L为高尿酸血症(hyperuricemia)。在血清尿酸浓度超过尿酸盐的溶解度时,就有可能尿酸钠的针状结晶沉淀于关节、肌腱、韧带、肾锥体的间质组织等软组织。足够数量的尿酸盐结晶可引起急性炎症反应,如沉积于关节腔,形成急性关节炎。这是由于尿酸盐结晶被白细胞吞噬后,破坏溶酶体膜,使膜内酶释放损伤白细胞和周围组织,引起关节炎症状。表现为关节剧烈疼痛,反复发作,此即痛风(gout)。沉积于软组织的结晶称为痛风石,周围发生炎症反应则构成痛风结节。原发痛风可由于遗传所致的核苷酸代谢中某一种酶表达异常所致,继发性痛风则继发于多种疾病。继发性病因可能有大量服用葡萄糖、果糖与甘露糖,使体内嘌呤合成增加;或多发性骨髓瘤、红细胞增多症、恶性贫血、牛皮癣和广泛转移的恶性肿瘤,使核蛋白转换率增快,而尿酸生成过多;或细胞毒药物或放射治疗时,核酸分解亢进使尿酸盐进一步增高。
先天性遗传原发通风病因是次黄嘌呤-鸟嘌呤磷酸核糖转移酶(HGPRT)缺失所致,部分缺失时,临床表现为尿酸过多的痛风特征;当完全缺失时,表现为高尿酸血症、精神发育迟缓等特征的自毁容貌综合征。
HGPRT蛋白质分子为相同亚基的四聚体,每个亚基由217个氨基酸残基组成。HG-PRT基因定位于X染色体长臂远端,因此该病表现为X连锁。HGPRT基因长约34kb,而成熟的mRNA的长度只有1.6kb,可见DNA分子内含有大量的内含子。HGPRT的突变基因及表达产物(mRNA,蛋白质)已被分离、研究,和血红蛋白一样有许多突变类型,如有的109位丝氨酸被亮氨酸代替,有的103位丝氨酸被精氨酸代替,还有报告基因发生缺失、重排的严重发病患者。1983年Wilson等发现一例DNA上的TagI限制性内切酶的切点发生突变,使在正常情况下可切出2.0kb片段的电泳区带,由于切点突变而失去酶切作用,变成4.0kb的区带出现。在蛋白质水平进一步研究发现50位的精氨酸被甘氨酸取代。
| 关于“临床生物化学/先天性代谢缺陷病”的留言: | |
|
目前暂无留言 | |
| 添加留言 | |