药理学/一级药动学指标间的相互关系
| 医学电子书 >> 《药理学》 >> 药物代谢动力学 >> 药物消除动力学 >> 一级药动学指标间的相互关系 |
| 药理学 |
|
|
1.F=A/D×100%口服剂量(D)由于不能100%吸收及存在首关消除效应,能进入体循环的药量(A)只占D的一部分,这就是生物利用度(F)。药动学计算时应采用绝对生物利用度,相对生物利用度作为评比药物制剂质量的指标。生物利用度还包括吸收速度问题,达峰时间(Tpeak)是一个参考指标。
2.A=C.Vd或C=A/Vd体内药量(A)与血药浓度(C)比值固定,在许多药动学公式中,A与C可以通用,如At=也可用Ct=。
3.Cp=[D]+[DP] 血浆中药物有游离型(D)与血浆蛋白结合型(DP),定量测定时需将血浆蛋白沉淀除去,故通常所说的血浆药物浓度(Cp)是指[D]与[DP]的总和。只有透析法或超离心法才可能将二者分离以计算药物的血浆蛋白结合率 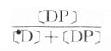 ×100% 。
×100% 。
4.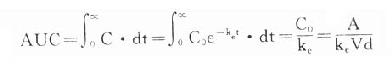 曲线下面积(AUC)是一个可用实验方法测定的药动学指标。它反映进入体循环药量的多少。时量曲线某一时间区段下的AUC反映该时间内的体内药量。AUC是独立于房室模型的药动学参数,常用于估算血浆清除率(Cl)。
曲线下面积(AUC)是一个可用实验方法测定的药动学指标。它反映进入体循环药量的多少。时量曲线某一时间区段下的AUC反映该时间内的体内药量。AUC是独立于房室模型的药动学参数,常用于估算血浆清除率(Cl)。
5.ke=0.693/t1/2=RE/A=CL/Vd 消除速率常数是药物瞬时消除的百分率而不是单位时间药物消除速率(RE),是决定t1/2的参数,但其本身又取决于Cl及Vd,故不是独立的药动学指标。
6.Vd=A/C0=A/AUC ke 表现分布容积(Vd)是独立的药动学指标,不是实际的体液容积,取决于药物在体液的分布。Vd大的药物与组织蛋白结合多,主要分布于细胞内液及组织间液。Vd小的药物与血浆蛋白结合多,较集中于血浆。Vd不因A多少而变化。
7.CL=keVd=RE/Cp=A/AUC 血浆清除率(Cl)是肝肾等清除率的总和,也不是实际的药物消除速率(RE),是另一个独立于A的重要药动学指标,但受肝肾功能的影响。
8.t1/2=0.693/ke=0.693Vd/CL 血浆药物消除半衰期(t1/2)是一个非常实用的药动学指标,虽然独立于A,但受Cl及Vd双重制约,Cl大时t1/2短,Vd大时t1/2长。例如庆大霉素Cl小(60ml.min-1), Vd也小(0.25L.kg-1),其t1/2不长(2~3h)。氯喹Cl大(700ml.min-1),Vd也大(185L.kg-1),其t1/2并不短(8天)。药物在吸收及分布过程中也有半衰期,分别用t1/2a及t1/2α表示。
9.稳态时RA=RE=CSS.Cl=CSS.Vd.ke
故 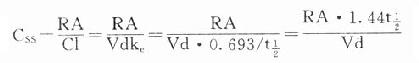 CSS是恒速连续给药达到稳态时平均血药浓度,应该和预期的有效浓度相等。必要时可以按达到的CSS与预期的CSS比值调整剂量或给药速度(RA)。
CSS是恒速连续给药达到稳态时平均血药浓度,应该和预期的有效浓度相等。必要时可以按达到的CSS与预期的CSS比值调整剂量或给药速度(RA)。
10. 分次定时定量给药时,CSS上下波动。当每t1/2给药一次时,其峰值(CSS- max)与谷值(CSS- min)的比值为2,缩短给药间隔可以减少CSS波动。
分次定时定量给药时,CSS上下波动。当每t1/2给药一次时,其峰值(CSS- max)与谷值(CSS- min)的比值为2,缩短给药间隔可以减少CSS波动。
11.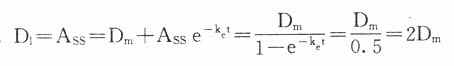 每t1/2给药一次时,首次给予加倍剂量,即负荷剂量(D1)可以立即达到CSS。
每t1/2给药一次时,首次给予加倍剂量,即负荷剂量(D1)可以立即达到CSS。
| 关于“药理学/一级药动学指标间的相互关系”的留言: | |
|
目前暂无留言 | |
| 添加留言 | |